How Analytics Are Transforming Pharmacovigilance
Within the last decade, there has been a growing awareness that the scope of Pharmacovigilance (PV) should be extended beyond the strict framework of detecting signals of safety concern. Nowadays, PV organizations face increasing pressures to enhance their analytic capabilities and become a value-added partner through the product development lifecycle. Increased regulatory scrutiny and greater emphasis on safety from consumers provide added pressure for companies to ensure that they are being proactive about accurately monitoring and assessing the benefit-risk profile of a medicinal product as early as possible in the product’s lifecycle. This is where pharmacovigilance analytics come into place.
Pharmacovigilance Analytics Defined
Pharmacovigilance analytics can be defined as the use of advanced analytic techniques with the purpose of examining large and varied data sets containing safety information, to uncover hidden patterns, unknown correlations, trends, patient preferences and other useful information that can help organizations make more-informed business decisions.
The effective management of safety data across multiple platforms is critical for the analysis and understanding of safety events. In an increasingly challenging business environment, pharmacovigilance analytics provides an opportunity to better utilize data, both to comply with regulatory authorities reporting requirements, and to drive actionable insights that can predict and prevent adverse events (AE).
Modern companies will use a value-based approach for pharmacovigilance analytics. This type of approach emphasizes quality and prevention over other aspects of the classical PV work. While the classical approach to AE analysis responds to Reporting (What happened?) and basic Monitoring (What is happening now?), we need to go beyond that, by enhancing our monitoring activities and being able to cover and respond to additional aspects like Evaluation (Why did it happen?), Prediction (What will happen?), and Prescription (To whom will it happen?).
Purpose of Pharmacovigilance Analytics
Our purpose should be to establish a PV data analytics process designed to leverage big data and the benefits of using such data across the value chain to build synergy between traditional (including regulatory obligations) analytics and big data analytics to provide faster and better insights to the organization.
Pharmacovigilance analytics serves as one of the instruments for the continuous monitoring of pharmacovigilance data. All available evidence on the benefit-risk balance of medicinal products and all their relevant aspects should be sought. All new information that could have an impact on the benefit-risk balance and the use of a product, should be considered for decision making.
Basically, in the framework of pharmacovigilance, PV analytics should be applied to gain insights by integrating data related to medicinal products from multiple sources and applying techniques to search, compare, and summarize them.
Overview of Pharmacovigilance Analytics
Pharmacovigilance departments must have in place the ability to quickly identify risks based on internal and external information, through processes that identify and extract product and indication-specific information from across the organization.
PV analytics will be used for, but not limited to:
- Monitoring of compliance regarding AE / case management
- Supporting analysis for signal detection
- Contributing to the elaboration of benefit-risk assessments (as stand-alone, or as part of regulatory aggregate reports), and
- Providing knowledge discovery on the factors governing the association between the exposure to a medicinal product and its effects on the population
The company will be able to leverage the knowledge discovery process and benefit of its results across the organization. For example, new insights can be used for the drug discovery process, and to prevent reputation and monetary loss from withdrawal of the medicinal product.
PV analytics uses data integration. The analysis of data integrated from multiple sources provides a synergy that generates real value, in contrast to multiple-step analyses that make it difficult to understand the big picture.
For that purpose, PV analytics applies new techniques for analysis of data including, but not limited to, data mining, text and information mining, and visualization tools. For more detailed information about analytical methods and tools click here.
Following we develop the main aspects of PV analytics enumerated in the Overview section.
Adverse Event / Case Management Compliance Monitoring
The biggest challenges facing pharmacovigilance are the rising and unpredictable AE case volumes, increasing complexity and cost, a lack of investment in new technologies (automation) and process improvements, as well as shortage of well-defined metrics. All these challenges can contribute to a general decrease in operational performance and ultimately case quality or compliance. To avoid and prevent these problems, companies are encouraged to set up an AE / Case Management compliance monitoring system.
Monitoring can be done by PV scientists, in collaboration with PV operations. They will monitor PV organization’s operational efficiency, including case processing, identification of issues in case workflow, contract research organization (CRO) management, and case processor management.
Proposed metrics for this section are:
- AE analytics
- Case processing metrics
- Case submission metrics
- Key performance indicators
- Trend analysis
Data sources: Safety Database
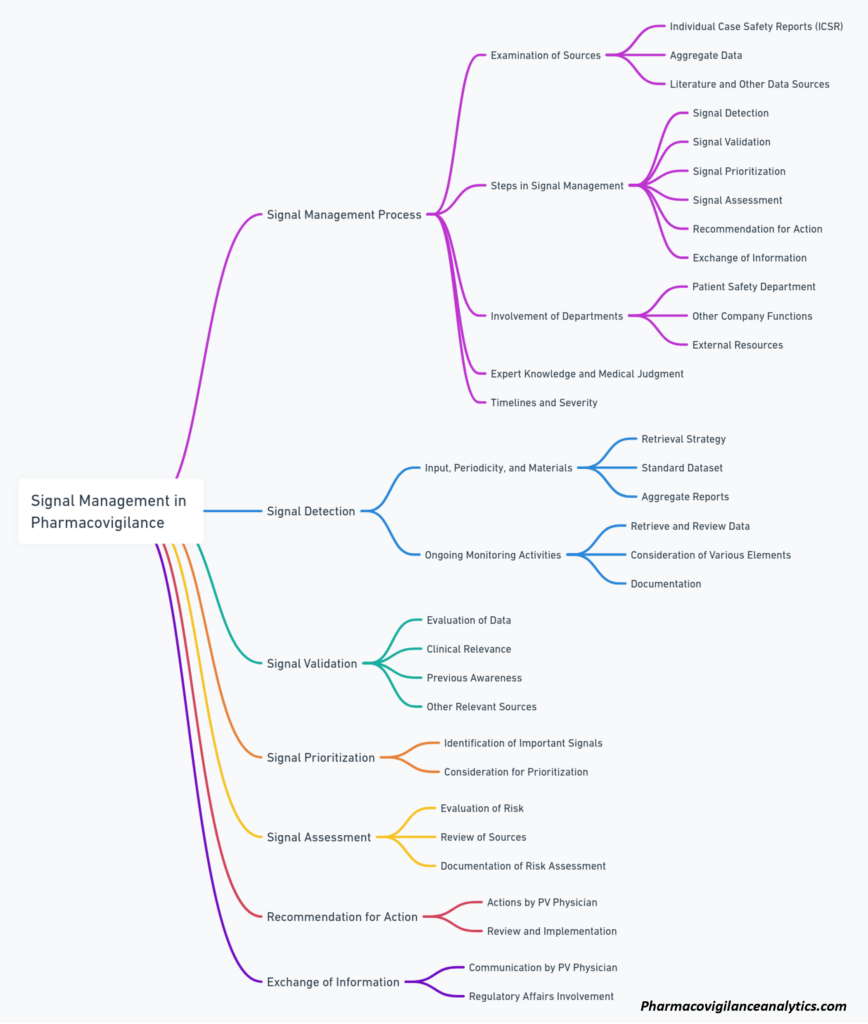
Signal Detection and Management Analytics
In accordance with processes governing Signal Detection and Management, PV scientist generates a monthly report for each company product. The report is delivered not later than 7 business days following the reporting month, and includes the following metrics for the reporting month, including cumulative statistics:
Proposed metrics, applicable to all AEs, highlighting Designated Medical Events (DME), and Targeted Medical Events (TME):
- Descriptive analysis of AEs by expectedness, causality, severity and outcome
- Reporting rates of AEs submitted, by geographic area, age, gender, and race, classified using MedDRA Preferred Term (PT) and System Organ Class (SOC)
- Proportional reporting ratio (PRR), and reporting odds ratio (ROR), including their statistical significance, calculated for each AE on a monthly basis and cumulative
- Trend analysis of the previous metrics
Data sources: a combination of safety database, FDA Adverse Event Reporting System (FAERS), EMA EudraVigilance, and WHO Vigibase
Active Benefit-Risk Identification and Analysis
Inspired in ARIA (Active Risk Identification and Analysis) model from FDA Sentinel Initiative. PV analytics will use an integrated, active benefit-risk identification and analysis system. This system will be comprised of pre-defined, parametrized, , and re-usable querying tools that will enable safety surveillance using company data platform, including medical operations, as well as commercial operations databases.
The objective of this part of the PV analytics operation is to take advantage of the enormous amount of healthcare data that is generated on daily basis. By using up-to-date analytic methods, PV analytics will be able to promptly identify emerging risks (and possibly benefits), as well as to acquire better insights on the safety profile of company medicinal products.
Pattern identification of product-event combinations, multivariable classification of risks, and the identification of factors associated with the risk of experiencing an AE, are among the main objectives of this section. This will allow the creation of models that are able to estimate the probability of an AE for a given group of patients, with the ultimate goal of utilizing this information for the prevention of such AEs.
Specifically, this section wants to provide analytical support for the benefit-risk assessment of medicinal products, being ad-hoc or to be added to regulatory safety reports requiring benefit-risk analysis.
PV analytics will create algorithms for use in administrative and clinical data environments to identify company-prioritized health outcomes that may be related to company medicinal products. Apart from all potential AEs company designated medical events and targeted medical events will be specifically monitored.
Queries will be created for, but not limited to:
- Calculation of event rates of exposure, outcomes and conditions
- Identification of the exposure of interest (company medicinal product, same-class products), and determination of the exposed time
- Identification of most frequently observed event codes
- Identification of the exposure and treatment patterns of the company medicinal products
- Characterization of concomitant medications
- Estimation of propensity scores following the identification of exposures, follow-up times, exposures and covariates
- Estimation of treatment effects, including hazard ratios and incidence rate differences
Active surveillance using sequential monitoring
- Given the longitudinal nature of the AE monitoring system, a specific type of statistical tools is required. One approach applied to new safety systems using electronic data to assess safety is sequential monitoring, which permits repeated estimation and testing of associations between a new medicinal product and potential AEs over time.
- Sequential analysis computes the test statistic at periodic time intervals as data accumulate, compares this test statistic to a prespecified signaling threshold, and stops if the observed test statistic is more extreme than the threshold. This way, sequential test can facilitate earlier identification of safety signals as soon as sufficient information from the electronic health care database becomes available to detect elevated AE risks.
- Although used extensively in clinical development, the application of sequential analysis to postmarket surveillance is relatively new. The following planning steps will be applied to safety evaluations in observational, electronic health-care database settings, either for a one-time analysis or multiple sequential analyses over time:
- Use available data (or existing literature) to conduct a feasibility assessment and prespecify the surveillance plan. Pre-specification of the surveillance design and analytical plan is critical.
- Describe uptake for the product of interest to determine if we will have enough sample size for the analysis. Use existing data to inform surveillance planning can reduce the number of assumptions that need to be made at the planning phase and, in turn, minimize downstream changes to initial sequential plans.
- Statistically evaluate, jointly select, and clearly communicate the final sequential design. Selection of a sequential design should include statistical evaluation and clear communication of the sequential design and analysis with all those designing and interpreting the safety surveillance activity so that the operating characteristics are well understood in advance of implementation.
- Finally, reports will be generated to reflect knowledge acquired on benefits and risks that appeared during the time window covered by the report. Ad-hoc reports will be created when needed.